All content on Eyewiki is protected by copyright law and the Terms of Service. This content may not be reproduced, copied, or put into any artificial intelligence program, including large language and generative AI models, without permission from the Academy.
A number of systemic metabolic disorders of genetic origin affect the anterior portion of the eye. Many of the corneal manifestations of systemic disease are alterations in corneal clarity and function caused by abnormal storage of metabolic substances, such as proteins, carbohydrates, and lipids.
Metabolic keratopathies (MK) are usually autosomal recessive and a single enzyme defect or absence often accounts for the clinical manifestations. Various gene loci have been determined along with its associated biochemical defect in patients with MK.
MK can be confused with corneal dystrophies as both are bilateral and are often associated with systemic involvement. The following points can be used to distinguish between the two:
MK affects the peripheral as well as the central cornea.
The corneal changes in metabolic disorders may involve more than one layer of the cornea.
There is typically progression over time in patients with MK.
Etiology
Metabolic keratopathies are not only genetically inherited but can also be acquired through prolonged metabolic dysregulation such as poorly managed diabetes. Chronic systemic inflammation, oxidative stress, and altered metabolic flux have emerged as unifying mechanisms underlying corneal manifestations in diverse metabolic disorders[1].
The clinical manifestations are dependent upon the enzymatic defects and accumulation of specific toxic metabolic substances. Patients can present with a wide range of symptoms (including ocular, cardiac, neurological deficits) and even death at an early age.
General Pathology
Enzymatic defects along the normal path of metabolism cause accumulation of toxic byproducts which circulate in the body. Depending upon the level of toxicity, the result may be disabling or even life-threatening.
Metabolic Disorders
Diabetes Mellitus
Epidemiology
Diabetes mellitus (DM) affects 451 million adults between the ages of 20 to 99 years of age worldwide. Diabetic keratopathy affects 50-70% of patients with DM.
Pathogenesis
Insulin deficiency and/or resistance
Enzymatic abnormalities
Increase in polyol metabolism
Increase in glycation of protein components
Increase in advanced glycation end products (AGE)
Structural & functional abnormalities
Basement membrane thickening
Decreased corneal nerve bundles
Loss of nerve-derived growth factors (IGF-1, substance P)
Treatment
Topical lubricants
Patching
Bandage contact lens
Topical growth factors (IGF-1 and Substance P and Epidermal Growth Factor)
Oral and topical aldose reductase inhibitor
Cenegermin
Neurotrophic peptides (e.g., Thymosin β4) and regenerating agents (RGTA)[2]
Promote corneal nerve regeneration and epithelial healing [2]
Disorders of lysosomal storage
Figure: Subtypes of MPS 1
Lysosomes are organelles that digest protein, nucleic acids, lipids, and carbohydrates.
A. Mucopolysaccharidoses (MPS)
MPS occurs with a prevalence of 1:10,000 births. Screening is performed by measuring urine glycosaminoglycans (GAGs). Diagnosis is based on blood assays.
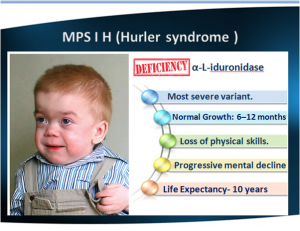
Hurler's is the most severe form. Hurler is characterized by severe mental impairment.
Scheie is the mildest form.
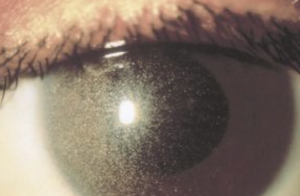
All 3 forms can present with severe corneal clouding that mostly involves the stroma (sparing the endothelium and Descemet's membrane.Figure: Hurler's Syndrome
2. MPS II (Hunter's syndrome)
Defective enzyme: iduronate-2-sulfatase
Accumulation: Dermatan & heparan sulfate (GAGs)
No to minimal corneal changes.
3. MPS III (Sanfilippo)
Defective enzyme: Deficiencies in 4 different enzymes have been identified, but are clinically indistinguishable
Accumulation: GAGs
Presents between age 2-6yrs
Rapid mental deterioration
No corneal changes, though mild to severe retinopathy can be detected
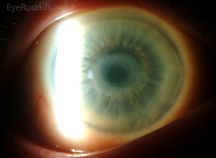
Anterior segment optical coherence tomography (AS-OCT) and confocal microscopy are now standard in tracking corneal crystal deposition and treatment response in cystinosis[12].
Subtype
Nephropathic (most common & severe form)
Cornea findings appear from the 1st year of life
Retinal depigmentation appears by 3-7 years of age
Glaucoma
Renal Fanconi syndrome, end-stage renal disease (ESRD) by 10 years of age
Intermediate (juvenile-onset or adolescent)
Corneal crystals are evident
No retinopathy
Slower progression to ESRD
Non-nephropathic aka benign or adult-type
Treatment
Renal transplantation may prolong life but does not decrease crystal accumulation
PKP, crystals can reappear in the corneal graft.
Cysteamine has revolutionized the treatment of cystinosis
Depletes cells of cystine (reacts with cystine, which allows it to leave the lysosome)
The systemic form can stabilize renal function and the retinopathy, but there is little effect on the corneal crystals
Topical cysteamine (0.55% cysteamine hydrochloride solution) 10-12 times a day has been shown to dissolve corneal crystals and improves ocular symptoms
A new cysteamine gel formulation with extended release is under trial, reducing dosing frequency[12].
Disorders of lipid & lipid metabolism
Characterized by premature onset of coronary artery disease & PVD
Tyrosine and phenylalanine restricted diet in infancy is the most effective therapy, which can clear the corneal lesions and ameliorate systemic symptoms
B. Alkaptonuria
Autosomal recessive
Enzyme deficiency: Homogentisic acid oxidase
Accumulation: Homogentisic acid (an intermediate breakdown product in the catabolism of phenylalanine and tyrosine)
Ocular findings
Blue-black pigmentation of the conjunctival and intrapalpebral scleral, usually at the insertion of the horizontal rectus muscles
Brownish-black “oil droplet” pigmentation of the cornea
Familial amyloidotic polyneuropathy type V or Meretoja syndrome
Rare dominantly inherited conditions
Appears in early adulthood
Rarely described in patients who are not Finnish
There is a similar condition that is nonfamilial
Mutation of GLN (gene located on chromosome 9) encoding the actin severing protein Gelsolin
Triad of symptoms
Ocular (LCD II, glaucoma)
Lattice, progressive loss of corneal sensory nerves, dry eyes
Epithelial erosions later in life
Visual acuity is usually retained until the 6th decade but visual loss after 60 years of age is common
Neurological (cranial, peripheral, and autonomic neuropathies)
Dermatological manifestations
Other symptoms
Renal and cardiac involvement
Treatment
PKP, can be complicated by neurotrophic persistent epithelial defect. Integrity is dependent on an intact nerve supply to the cornea which is absent in amyloidosis type V
Excimer laser phototherapeutic keratectomy has been successful in patients who have had recurrences after lamellar keratoplasty
3. Localized secondary corneal amyloidosis
Localized, follows chronic diseases such as neoplasms, infections, connective tissue disorders, and trauma4.
4. Secondary systemic amyloidosis with corneal manifestations
Myeloproliferative disorders such as monoclonal gammopathy
Heavy chain amyloidosis
D. Gout
Accumulation of urea (urate crystals) in tissue
Most common in middle aged men
Etiologies
Myeloproliferative disorder
Alcohol
Chemotherapy
Obesity
Ocular findings
Urate keratopathy- chalky deposits or fine refractile crystals in the corneal stroma
↑Sapra, B., Mahajan, D., & Chaudhary, S. (2022). Eye in metabolic disorders: Manifestations and drug delivery systems. In Novel Drug Delivery Systems for Metabolic Disorders (Elsevier). Link
↑ 2.02.1Syed, Z. A., Meghpara, B. B., & Hammersmith, K. M. (2021). Corneal manifestations of metabolic disease. In Albert and Jakobiec’s Principles and Practice of Ophthalmology. Link (Springer)
↑Ramani PK, Parayil Sankaran B. Tay-Sachs Disease. [Updated 2023 Jan 25]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2023 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK564432/
↑Sandhoff Disease. National Institute of Neurological Disorders and Stroke. https://www.ninds.nih.gov/health-information/disorders/sandhoff-disease. Accessed [July 3, 2023]
↑Xiao C, Toro C, Tifft C. GM2 Activator Deficiency. 2022 Aug 25. In: Adam MP, Mirzaa GM, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2023. Available from: https://www.ncbi.nlm.nih.gov/books/NBK583219/
↑Tripathy K, Patel BC. Cherry Red Spot. [Updated 2023 Apr 3]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2023 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK539841/
↑Bajwa H, Azhar W. Niemann-Pick Disease. [Updated 2023 Mar 6]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2023 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK556129/
↑ 12.012.1Rohrbach, J. M., & Seitz, B. (2024). Die Hornhaut als Indikator für Systemerkrankungen. In Die Augenheilkunde: Das Referenzwerk. Springer. PDF
Krachmer J, Mannis M, Holland E: CORNEA, 2nd ed.Elsevier Mosby, 2017, 620-644.
Priyadarsini S, Whelchel A, Nicholas S, Sharif R, Riaz K, Karamichos D. Diabetic Keratopathy: Insights and Challenges. Surv Ophthalmol 2020.
External Disease and Cornea, Section 8. Basic and Clinical Science Course, AAO, 2006./li>
The Academy uses cookies to analyze performance and provide relevant personalized content to users of our website.