All content on Eyewiki is protected by copyright law and the Terms of Service. This content may not be reproduced, copied, or put into any artificial intelligence program, including large language and generative AI models, without permission from the Academy.
Epilepsy is a condition characterized by recurrent, unprovoked seizures, which are episodes of abnormally hypersynchronous brain activity. Seizures are classified by their onset: focal, in which only one cerebral hemisphere is affected; generalized, in which both hemispheres are affected; and unknown onset per the 2017 International League Against Epilepsy (ILAE) classification.[1] Epilepsy is categorized into four main types: focal, generalized, combined generalized and focal, and unknown.[1] Epilepsy types exist on a wide spectrum and often may present differently from person-to-person. During seizure episodes, people can experience auras, muscle jerking, automatisms, altered level of consciousness and convulsions.[1][2] Some characteristic ophthalmologic signs can also be observed including visual hallucinations, illusions, visual field loss, eye deviation, nystagmus, eyelid automatism and myoclonia.[2] These observed ophthalmologic manifestations may be seen in epilepsy and will be discussed in this article.
Etiology
The cause of epilepsy has not been definitively discerned and can be broadly categorized as structural, genetic, infectious, metabolic, immune, or unknown (Table 1).[1]
Etiologies are not necessarily mutually exclusive and often happen in conjunction. For example, a structural abnormality may be genetically inherited, so the etiology may be both structural and genetic.[1] Epileptogenesis is the term used to describe how epilepsy develops. Usually, epileptogenesis involves some type of neurotransmitter imbalance (GABA, glutamate) with abnormal neuronal excitation and excessive synchronization.[3]
Table 1. Examples of different etiologies of epilepsy. KNCQ2 = potassium voltage-gated channel subfamily Q member 2, BFGE = benign familial genetic epilepsy, SCN1a = sodium voltage-gated channel alpha subunit 1, CAE = childhood absence epilepsy, JME = juvenile myoclonic epilepsy, GEFS+ = generalized epilepsy with febrile seizures plus, TB = tuberculosis, HIV = human immunodeficiency virus, CMV = cytomegalovirus, NMDA = N-methyl-D-aspartate, LGI1 = leucine-rich glioma inactivated 1 [1][4][5]
Risk Factors
The risk factors for epilepsy can include: stroke, family history of epilepsy, head trauma, CNS infection.[6]
Pathophysiology
The pathophysiology of epilepsy is not fully understood. Different types of epilepsy may result from abnormal patters of neuronal migration, cortical abnormalities resulting in hyperexcitability, periventricular heterotopia, increased plasma glutamate levels, focal abnormalities, mutated voltage-gated Na+ and K+ channels, temporal hippocampus atrophy, amygdala kindling, neurotransmitter imbalance (GABA, glutamate).[3]
Diagnosis
To make a diagnosis of epilepsy, a neurologist may do a full neurological exam, conduct blood tests, monitor EEG activity, and obtain neuroimaging (CT/MRI).
The clinical diagnosis of epilepsy is defined by at least two unprovoked seizures occurring more than 24 hours apart, one unprovoked seizure with risk of future seizure, or diagnosis of epilepsy syndrome.[7]
Neurological Manifestations
Auras
Precedes a generalized seizure
Typically associated with temporal lobe epilepsy[4]
May include motor, sensory, autonomic or psychic manifestations
Automatisms
Unconscious, automatic behaviors that may occur in complex seizures can include:
Complexity of hallucinations can be caused by ictal spread to the temporal lobes[2]
Figure 1. Visual aura. It can start as a scotoma that expands, then dissipates. Timestamps show the passage of time in minutes. Image obtained from AAO.org.
Can lead to autoscopy, which is a depersonalization where people perceive their body image from an external perspective[2]
“Alice in Wonderland” syndrome – kinetopsia, complex hallucinations, somatic hallucinations of body parts (e.g., aschematia), stationary object moving away from observer (e.g., porropsia), depersonalization, out-of body experiences[10]
Tonic eye deviation
May be associated with versive movements and may occur due to excessive activation of the contralateral frontal eye field[2]
Ictal spread preceding generalization of seizures is a possible mechanism for the eye deviation[2]
Photosensitivity
Some people with reflex epilepsy may experience photosensitivity and can exhibit paroxysms on EEG when eyes are closed[11]
Photic stimulation for extended period of time may trigger a seizure and is presumed to be due to decreased GABAergic inhibition[11]
Nystagmus
Epileptic nystagmus is not a commonly observed sign in patients, but it can be caused by focal onset seizures and contains a fast and slow phase[2]
Fast phases are triggered by the contralateral saccadic gaze centers[2]
Palinopsia
Persistent appearance of visual images following removal of the stimulus, also known as visual perseveration[12]
May be caused by seizures of the visual processing center in the brain
EEG evidence of posterior temporal lesion associated with epilepsy and palinopsia[12]
Eyelid myoclonia
Forceful twitches of the eyelid that may include the eyebrows[2]
Eyelid flutter
Repeated eye blinking seen in different forms of epilepsy, can be seen in occipital/temporal/frontal lobe seizures[2]
Unilateral presentation can predict ipsilateral cortical pathology[2]
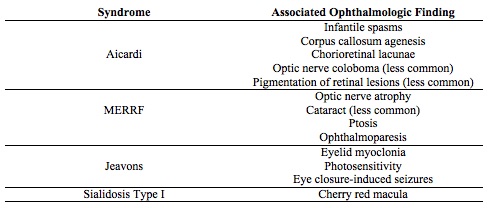
Select epileptic syndromes with specific ophthalmic findings
AICARDI SYNDROME
Aicardi syndrome is an X-linked recessive disorder that involves corpus callosum agenesis and chorioretinal lacunae.[13][14] Typically, only females are affected, as the mutation is lethal to males in utero.
Aicardi may show asynchronous burst-suppression on EEG. Clinically, a diagnosis can be made following observation of a characteristic triad: infantile spasms, chorioretinal lacunae (Figure 2), and callosal agenesis.[14] Optic nerve coloboma, and pigmentation of retinal lesions can also be observed.[13]
Aicardi syndrome can often present with intractable seizures with limited control. The treatment includes anti-epileptic drugs (AED) but with limited success.[13] Mental retardation is typically seen, with only a 40% survival rate by age 14.[14]
MERRF has a clinically diagnostic myoclonus,[15][16] and a genetic mutation most frequently found at nucleotide pair 8334 is diagnostic of MERRF.[16][17]
MERRF is a mitochondrial disorder; the exact mechanism of how mutations in this gene of mitochondrial DNA cause the clinical symptoms observed in MERRF is unclear.
MERRF has characteristic optic nerve atrophy[15] and in some cases, cataract formation, ptosis and ophthalmoparesis.[16]
JEAVONS SYNDROME
Jeavons syndrome is considered an idiopathic generalized epileptic syndrome and has a characteristic triad: eyelid myoclonia, eye-closure induced seizures, and photosensitivity.[18] Eyelid myolonia has a jerking of eyelids and upward deviation with irregular EEG following eye-closure.[19]
Jeavons syndrome may cause occipital lobe seizures. Upon exposure to light, the volume of the occipital cortex changes. If the visual stimuli is intense enough, the epileptic cortex in the occipital cortex can be activated and lead to eyelid myoclonia.[20]
The eyelid myoclonia found in Jeavons is resistant to AED, but AED need to be used in people with photosensitivity.[19]
Photosensitivity typically decreases with aging in Jeavons syndrome, while eyelid myoclonia persists.[18]
SIALIDOSIS TYPE I
Sialidosis type I involves an autosomal recessive mutation in NEU1 on chromosome 6p21.3.[21][22] Sialidase deficiency leads to defects in lysosomal storage, particularly affecting the activation of macrophages and T-lymphocytes.[22]
The onset is juvenile or young adult, usually manifesting in the second decade of life.
Sialidosis type I is diagnosed by the presence of sialic acid in urine and a neuraminidase enzyme deficiency in fibroblasts, a normal EEG, and enzyme assays to measure activity in blood leukocytes.[21][22]
Sialidosis type I is associated with myoclonus. A cherry red macula is pathognomonic, and sometimes, cataracts in young patients are seen .[21]
In Sialidosis type I, valproate is generally the first-line drug[21], with enzyme replacement therapy, bone marrow transplantation, and gene therapy showing some benefit in mouse models.[22] However, enzyme-replacement has its own problems of inability to cross the blood-brain barrier and causes anaphylaxis reactions.
Table 2. A selection of epileptic syndromes with characteristic ophthalmologic findings. [14][15][16][17][20][18][19][21]Sialidosis type I can lead to progressive degeneration of multiple systems: bone, nervous, musculoskeletal, and immune.[22] Within a few years, patients can become wheelchair-bound.[21]
Anti-epileptic drugs associated with ophthalmic findings
Please refer to the upcoming Eyewiki article entitled “Ophthalmic Manifestations associated with Anti-Epileptic Drugs” for further details on this topic.
Differential Diagnosis
Migraine
Pathophysiology: Propagation of depolarization across the cerebral cortex known as cortical spreading depression, unlike hypersynchronous discharge and changing ion channel permeability seen in epilepsy[23]
Visual hallucinations in migraine may last hours with fortification and march and build-up patterns, while those found in epilepsy may only last for a few seconds[2]
Transient ischemic attack
Typically negative symptoms such as numbness and monocular visual loss, compared to positive symptoms of seizures like automatisms and muscle jerking[24]
Positive visual symptoms may occur with ischemia. Examples include homonymous field defect if occipital ischemia occurs and penduncular hallucinosis in the setting of thalamic, midbrain or basal ganglia ischemia[2] [Basal ganglia]
Time scale of recurrence may be days to weeks, compared to seizures which may recur yearly[24]
Rare for loss of consciousness[24], rare for Jacksonian march[2][25], which begins with twitching in a finger, toe, or mouth and progressing to the entire ipsilateral hand, foot, and face.[26]
Management
General Treatment
Epilepsy can be treated with AED such as carbamazepine, ethosuximide, oxcarbazepine, valproate, leviracetam, and lamotrigine. Choice of drug is partially dependent on syndrome classification, and AED may not always successful in treating epilepsy.[27] Multi-drug regimen may often be employed. Recently, cannabidiol (CBD) has shown some efficacy as an adjuvant treatment in drug-resistant epilepsy and can help reduce seizure frequency.[28] Alternative treatment methods may include vagal nerve stimulation (VNS) and responsive focal cortical stimulation. VNS involves a less invasive surgery, where a device is implanted in the upper chest and cervical area that stimulates the vagus nerve in order to suppress seizure activity in the brain.[29] It can be a successful alternative to intracranial surgery.[29]Responsive focal cortical stimulation has been shown to have some success in treating medically refractory epilepsy.[30]
Prognosis
Epilepsy can be treated well with AED in some patients. Seizure frequency can be controlled with medications, but it may vary between patients.[27]
In patients who undergo a temporal lobectomy for their epilepsy, a homonymous hemianopsia can be observed post-surgery.[9]
References
↑ 1.01.11.21.31.41.5Scheffer IE, Berkovic S, Capovilla G, et al. ILAE classification of the epilepsies: position paper of the ILAE Commission for Classification and Terminology. Epilepsia 2017;58(4):512-521.
↑Fisher RS, Acevedo C, Arzimanoglou A, et al. A practical clnical definition of epilepsy. Epilepsia 2014;55(4): 475-482.
↑ 8.08.18.28.3Hamer HM, Wyllie E, Luders HO, et al. Symptomatology of epileptic seizures in the first three years of life. Epilepsia 1999;40(7):837-844.
↑ 9.09.19.29.39.4Jensen I, Seedorff HH. Temporal lobe epilepsy and neuro-ophthalmology. Acta Ophthalmologica 1976;54:827-841.
↑Mastria G, Mancini V, Vigano A, Di Piero V. Alice in Wonderland Syndrome: a clinical and pathophysiologal review. Biomed Research International 2016;2016:82423145.
↑ 11.011.1Okudan ZV, Ozkara C. Reflex epilepsy: triggers and management strategies. Neuropsychiatric Disease and Treatment 2018;14:327-337.
↑ 13.013.113.2Ospina LH, Nayak H, McCormick AQ. Progressive pigmentation of chorioretinal lesions in Aicardi syndrome. Arch Ophthalmol 2004;122:790.
↑ 14.014.114.214.3Aicardi J. Aicardi syndrome. Brain & Development 2005;27:164-171.
↑ 15.015.115.2DiMauro S, Hirano M. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A, editors. SourceGeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 2003;1993-2018.
↑ 16.016.116.216.3Mancuso M, Orsucci D, Angelini C, et al. Myoclonus in mitochondrial disorders. Movement Disorders 2014;29(6):722-728.
↑ 17.017.1Shoffner JM, Lott MT, Lezza AMS, et al. Myoclonic epilepsy and ragged-red fiber disease (MERRF) is associated with a mitochondrial DNA tRNA-Lys mutation. Cell 1990;61:931-937.
↑ 18.018.118.2Abanoz YG, Abanoz Y, Ozkara C. Ongoing photosensitivity in an elderly patient with Jeavons syndrome. The Neurologist 2018;23:94-97.
↑ 19.019.119.2Covanis A. Photosensitivity in idiopathic generalized epilepsies. Epilepsia 2005;46(9):67-72.
↑ 20.020.1Viravan S, Go C, Ochi A, et al. Jeavons syndrome existing as occipital cortex initiating generalized epilepsy. Epilepsia 2011;52(7):1273-1279.
↑ 21.021.121.221.321.421.5Franceschetti S, Canafoglia L. Sialidoses. Epileptic Disord 2016;18(2):S89-S93.
↑ 22.022.122.222.322.4Igdoura SA. 2010. Sialidosis. In: Encyclopedia of Movement Disorders. Kompoliti K, and Verhagen Metman L (eds.) vol. 3, pp.114. Oxford: Academic Press.
↑Kim DW, Lee SK. Headache and epilepsy. Journal of Epilepsy Research 2017;7(1):7-15.
↑ 24.024.124.224 Nadarajan V, Perry RJ, Johnson J, Werring DJ. Transient ischemic attacks: mimics and chameleons. Pract Neurol 2014;14:23-31.